Sickle Cell Anemia: Understanding the Genetic Disorder and Its Impact on Health
Sickle cell anemia is a genetic disorder that affects millions of people worldwide. It is a form of sickle cell disease (SCD), a group of inherited red blood cell disorders. This condition is characterized by the presence of abnormal hemoglobin, known as hemoglobin S (HbS), which causes red blood cells to become rigid, sticky, and crescent-shaped. These misshapen cells can block blood flow, leading to a host of complications, including pain, infections, and organ damage. In this comprehensive article, we will delve into the causes, symptoms, diagnosis, treatment, and management of sickle cell anemia, as well as its impact on patients’ lives and the latest advancements in research.
What is Sickle Cell Anemia?
Sickle cell anemia is an autosomal recessive disorder, meaning that a person must inherit two copies of the defective gene, one from each parent, to develop the disease. The gene responsible for sickle cell anemia is the HBB gene, which provides instructions for making beta-globin, a component of hemoglobin. A mutation in this gene leads to the production of abnormal hemoglobin, known as hemoglobin S (HbS).
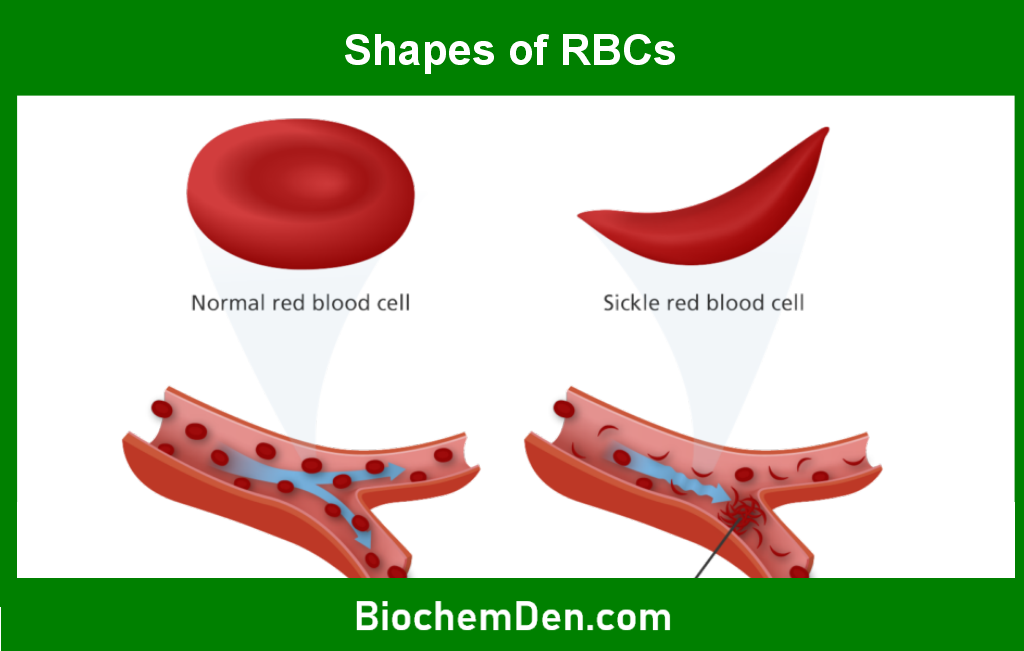
When oxygen levels are low, hemoglobin S causes red blood cells to assume a sickle shape. These sickle cells are less flexible and can stick to blood vessel walls, causing blockages that impede blood flow. This can result in tissue damage, pain, and other complications.
Epidemiology of Sickle Cell Anemia
Sickle cell anemia is most prevalent in populations with a high incidence of malaria, particularly in sub-Saharan Africa, the Middle East, India, and the Mediterranean region. This is because carriers of the sickle cell trait (those with one normal gene and one mutated gene) have a survival advantage against malaria. According to the World Health Organization (WHO), approximately 5% of the world’s population carries traits for hemoglobin disorders, with sickle cell anemia being the most common.
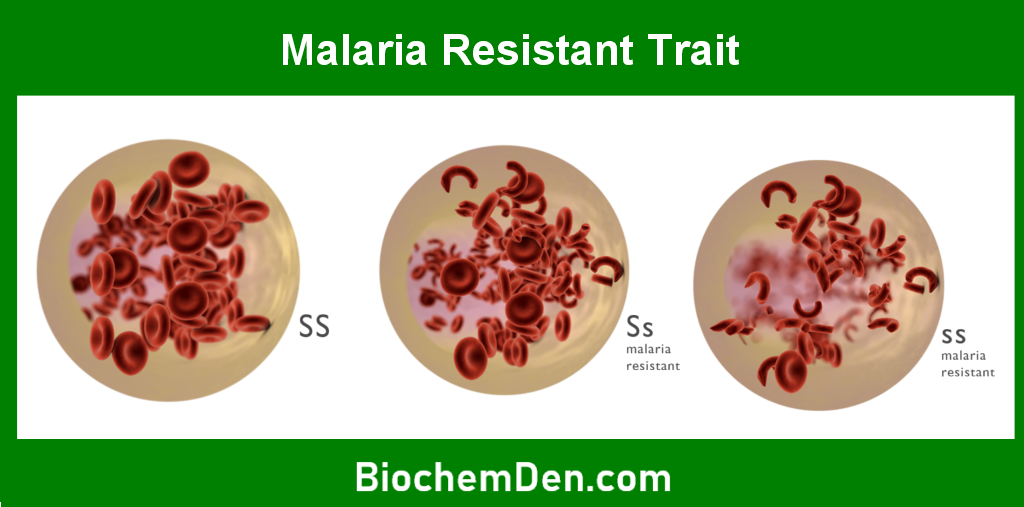
In the United States, sickle cell anemia affects about 100,000 people, primarily African Americans. It is estimated that 1 in 365 African American births and 1 in 16,300 Hispanic American births result in sickle cell anemia.
Common Symptoms of Sickle Cell Anemia
The symptoms of sickle cell anemia can vary widely in severity and frequency. Some individuals may experience mild symptoms, while others may suffer from severe, life-threatening complications. Common symptoms include:
- Anemia: Sickle cells are fragile and break apart easily, leading to a shortage of red blood cells. This results in anemia, which can cause fatigue, paleness, and shortness of breath.
- Pain Crises: Also known as vaso-occlusive crises, these episodes occur when sickle cells block blood flow to tissues and organs. Pain crises can last for hours to days and may require hospitalization.
- Swelling of Hands and Feet: Known as dactylitis, this occurs when sickle cells block blood flow to the extremities.
- Frequent Infections: Sickle cell anemia can damage the spleen, making patients more susceptible to infections, particularly from bacteria such as Streptococcus pneumoniae.
- Delayed Growth: Children with sickle cell anemia may experience delayed growth and puberty due to the chronic nature of the disease.
Complications of of Sickle Cell Anemia
Sickle cell anemia can lead to a range of complications, some of which can be life-threatening. These include:
- Acute Chest Syndrome: This is a serious condition characterized by chest pain, fever, and difficulty breathing. It occurs when sickle cells block blood flow in the lungs.
- Stroke: Sickle cells can block blood flow to the brain, leading to a stroke. This is more common in children with sickle cell anemia.
- Organ Damage: Chronic blockage of blood flow can damage organs such as the liver, kidneys, and spleen. This can lead to organ failure over time.
- Pulmonary Hypertension: High blood pressure in the lungs can develop as a result of chronic anemia and reduced oxygen levels.
- Leg Ulcers: Poor circulation can lead to the development of painful ulcers on the legs.
- Priapism: This is a painful, prolonged erection that can occur in males with sickle cell anemia due to blocked blood flow in the penis.
Diagnosis of Sickle Cell Anemia
a. Newborn Screening
In many countries, including the United States, newborn screening programs include tests for sickle cell anemia. Early diagnosis is crucial for managing the disease and preventing complications. The screening typically involves a blood test that checks for the presence of hemoglobin S.
b. Diagnostic Tests
If sickle cell anemia is suspected based on symptoms or family history, several diagnostic tests can confirm the diagnosis:
- Hemoglobin Electrophoresis: This test separates different types of hemoglobin in the blood and can identify the presence of hemoglobin S.
- Complete Blood Count (CBC): A CBC measures the number of red blood cells, white blood cells, and platelets in the blood. It can also detect anemia.
- Peripheral Blood Smear: A blood sample is examined under a microscope to check for the presence of sickle-shaped red blood cells.
- Genetic Testing: This test can identify mutations in the HBB gene and confirm a diagnosis of sickle cell anemia.
Treatment and Management of Sickle Cell Anemia
a. Medications
While there is no cure for sickle cell anemia, several treatments can help manage symptoms and reduce complications:
- Hydroxyurea: This medication can reduce the frequency of pain crises and the need for blood transfusions. It works by increasing the production of fetal hemoglobin, which can prevent the formation of sickle cells.
- Pain Relievers: Over-the-counter pain relievers such as acetaminophen and nonsteroidal anti-inflammatory drugs (NSAIDs) can help manage mild pain. For severe pain, opioids may be prescribed.
- Antibiotics: Children with sickle cell anemia are often prescribed penicillin to prevent infections, particularly from Streptococcus pneumoniae.
- L-Glutamine Oral Powder: This medication has been approved by the FDA to reduce the frequency of pain crises in patients with sickle cell anemia.
b. Blood Transfusions
Regular blood transfusions can help reduce the risk of stroke and other complications by increasing the number of normal red blood cells in the bloodstream.

However, frequent transfusions can lead to iron overload, which requires treatment with chelation therapy to remove excess iron from the body.
c. Bone Marrow Transplant
A bone marrow transplant, also known as a stem cell transplant, is the only cure for sickle cell anemia. This procedure involves replacing the patient’s bone marrow with healthy bone marrow from a compatible donor.
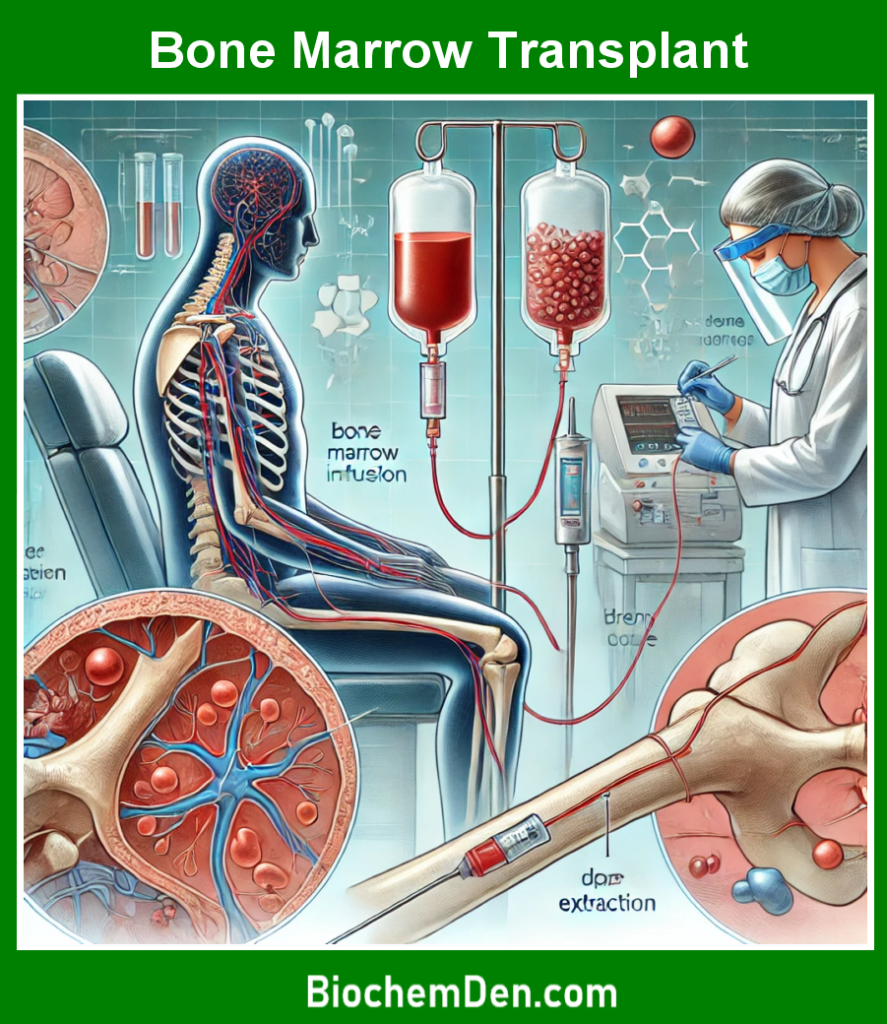
However, this treatment is not without risks, including the possibility of graft-versus-host disease, and is typically reserved for severe cases.
d. Gene Therapy
Gene therapy is an emerging treatment option for sickle cell anemia. This experimental approach involves modifying the patient’s own stem cells to produce normal hemoglobin.

While still in the experimental stages, early results have been promising, and gene therapy holds the potential for a cure without the risks associated with bone marrow transplants.
e. Lifestyle and Home Remedies
In addition to medical treatments, certain lifestyle changes and home remedies can help manage sickle cell anemia:
- Stay Hydrated: Drinking plenty of fluids can help prevent sickle cell crises by keeping the blood from becoming too viscous.
- Avoid Extreme Temperatures: Exposure to extreme heat or cold can trigger pain crises. It’s important to dress appropriately and avoid sudden temperature changes.
- Exercise Regularly: Moderate exercise can improve overall health and reduce the risk of complications. However, it’s important to avoid overexertion.
- Eat a Healthy Diet: A balanced diet rich in fruits, vegetables, and whole grains can help support overall health and prevent complications.
- Get Regular Check-ups: Regular visits to a healthcare provider can help monitor the disease and catch complications early.
Living with Sickle Cell Anemia
a. Emotional and Psychological Impact
Living with sickle cell anemia can be challenging, not only physically but also emotionally and psychologically. The chronic pain, frequent hospitalizations, and risk of complications can take a toll on mental health. Patients may experience anxiety, depression, and feelings of isolation. It’s important for patients to seek support from mental health professionals, support groups, and loved ones.
b. Impact on Quality of Life
Sickle cell anemia can significantly impact a patient’s quality of life. The disease can interfere with school, work, and social activities. Children with sickle cell anemia may miss school frequently due to pain crises and hospitalizations, while adults may struggle to maintain employment. It’s important for patients to work with their healthcare providers to develop a management plan that allows them to lead as normal a life as possible.
c. Support for Patients and Families
Support from family, friends, and the community is crucial for patients with sickle cell anemia. Support groups can provide a safe space for patients and families to share their experiences, offer advice, and find emotional support. Organizations such as the Sickle Cell Disease Association of America (SCDAA) and the Sickle Cell Society offer resources, advocacy, and support for patients and their families.
Advances in Sickle Cell Anemia Research
a. Gene Editing
One of the most exciting areas of research in sickle cell anemia is gene editing. Techniques such as CRISPR-Cas9 have shown promise in correcting the genetic mutation responsible for sickle cell anemia. Early clinical trials have demonstrated the potential to cure the disease by editing the patient’s own stem cells to produce normal hemoglobin.
b. New Medications
Researchers are continually working to develop new medications to treat sickle cell anemia. Some of the promising drugs in development include:
- Voxelotor: This medication works by preventing red blood cells from sickling and has shown promise in improving anemia and reducing the frequency of pain crises.
- Crizanlizumab: This drug is designed to prevent blood cells from sticking to blood vessel walls, reducing the risk of pain crises.
c. Stem Cell Research
Stem cell research holds the potential to revolutionize the treatment of sickle cell anemia. Scientists are exploring ways to use stem cells to repair damaged tissues and organs, as well as to develop new treatments that can cure the disease.
d. Global Initiatives
Global initiatives are underway to improve the diagnosis, treatment, and management of sickle cell anemia, particularly in low-resource settings. Organizations such as the WHO and the Global Sickle Cell Disease Network are working to increase awareness, improve access to care, and support research efforts.
Conclusion
Sickle cell anemia is a complex and challenging genetic disorder that affects millions of people worldwide. While there is currently no cure, advances in medical research and treatment options offer hope for patients and their families. Early diagnosis, comprehensive care, and ongoing support are essential for managing the disease and improving quality of life. As research continues to uncover new treatments and potential cures, the future looks promising for those living with sickle cell anemia.
By raising awareness, supporting research, and providing comprehensive care, we can work towards a world where sickle cell anemia no longer poses a significant threat to health and well-being. Whether you are a patient, caregiver, or healthcare provider, understanding the complexities of sickle cell anemia is the first step towards making a positive impact in the lives of those affected by this condition.
Frequently Asked Questions (FAQs) About Sickle Cell Anemia
Sickle cell anemia is a complex genetic disorder that raises many questions for patients, caregivers, and the general public.
What causes sickle cell anemia?
Sickle cell anemia is caused by a mutation in the HBB gene, which provides instructions for making beta-globin, a component of hemoglobin. This mutation leads to the production of hemoglobin S instead of normal hemoglobin (hemoglobin A). When oxygen levels are low, hemoglobin S causes red blood cells to sickle, leading to blockages in blood vessels and reduced oxygen delivery to tissues. The condition is inherited, meaning it is passed down from parents to their children. If both parents carry the sickle cell trait (one normal gene and one mutated gene), there is a 25% chance their child will have sickle cell anemia.
What are the symptoms of sickle cell anemia?
The symptoms of sickle cell anemia vary widely but often include chronic anemia, fatigue, and episodes of severe pain called pain crises or vaso-occlusive crises. Other common symptoms include swelling in the hands and feet (dactylitis), frequent infections, delayed growth in children, and jaundice (yellowing of the skin and eyes). Complications such as acute chest syndrome, stroke, and organ damage can also occur. Symptoms may range from mild to severe and can significantly impact a person’s quality of life.
How is sickle cell anemia diagnosed?
Sickle cell anemia is typically diagnosed through newborn screening programs, which use a blood test to detect the presence of hemoglobin S. If sickle cell anemia is suspected later in life, diagnostic tests such as hemoglobin electrophoresis, complete blood count (CBC), and genetic testing can confirm the diagnosis. Hemoglobin electrophoresis separates different types of hemoglobin in the blood, while genetic testing identifies mutations in the HBB gene. Early diagnosis is crucial for managing the disease and preventing complications.
Is there a cure for sickle cell anemia?
The only known cure for sickle cell anemia is a bone marrow transplant (also called a stem cell transplant), which involves replacing the patient’s bone marrow with healthy bone marrow from a compatible donor. However, this procedure carries significant risks, including graft-versus-host disease, and is not suitable for all patients. Other treatments, such as gene therapy, are currently being researched and show promise as potential cures. Most patients rely on treatments that manage symptoms and prevent complications, such as hydroxyurea, blood transfusions, and pain management.
What is a pain crisis, and how is it managed?
A pain crisis, also known as a vaso-occlusive crisis, occurs when sickle-shaped red blood cells block blood flow to tissues and organs, causing severe pain. These episodes can last for hours to days and often require hospitalization. Pain crises are managed with pain relievers, including over-the-counter medications like acetaminophen and NSAIDs for mild pain and opioids for severe pain. Hydration, warmth, and rest can also help alleviate symptoms. Preventive measures, such as taking hydroxyurea or avoiding triggers like dehydration and extreme temperatures, can reduce the frequency of pain crises.
How does sickle cell anemia affect daily life?
Sickle cell anemia can significantly impact daily life due to chronic pain, fatigue, and the risk of complications. Children may miss school frequently due to pain crises or hospitalizations, while adults may struggle to maintain employment. The emotional and psychological toll of living with a chronic illness can also lead to anxiety, depression, and feelings of isolation. However, with proper management, including medications, regular check-ups, and a healthy lifestyle, many individuals with sickle cell anemia can lead fulfilling lives.
Can sickle cell anemia be prevented?
Sickle cell anemia cannot be prevented because it is a genetic disorder. However, genetic counseling can help individuals understand their risk of passing the condition to their children. If both parents carry the sickle cell trait, there is a 25% chance their child will have sickle cell anemia. Prenatal testing can also determine if a fetus has the condition. Public health initiatives, such as newborn screening and education about the disease, play a crucial role in early diagnosis and management.
What are the long-term complications of sickle cell anemia?
Long-term complications of sickle cell anemia include organ damage, stroke, pulmonary hypertension, leg ulcers, and vision problems. Chronic blockages of blood flow can damage organs such as the liver, kidneys, and spleen, leading to organ failure over time. Patients are also at higher risk of infections due to spleen damage. Regular monitoring and preventive care, such as vaccinations and blood transfusions, can help reduce the risk of these complications.
What advancements are being made in sickle cell anemia research?
Significant advancements are being made in sickle cell anemia research, particularly in the areas of gene therapy and gene editing. Techniques like CRISPR-Cas9 are being explored to correct the genetic mutation responsible for the disease. New medications, such as voxelotor and crizanlizumab, are showing promise in reducing symptoms and complications. Additionally, global initiatives are working to improve access to care and support research efforts, particularly in low-resource settings. These advancements offer hope for more effective treatments and potentially a cure in the future.
Discover more from Biochemistry Den
Subscribe to get the latest posts sent to your email.




