Protein Folding: The Intricate Process Behind Protein Structure and Function
Proteins are indispensable macromolecules that carry out a vast array of biological functions within living organisms. From catalyzing metabolic reactions to fighting infections, proteins are involved in virtually every physiological process.
But how do these complex molecules carry out their diverse functions so effectively? The answer lies in protein folding, the physical process by which a protein chain acquires its unique 3D structure.
Protein folding is a critical process in the complex biology of the cell, as it determines the final three-dimensional shape of a protein.
Understanding this intricate mechanism involves studying the various stages of protein folding. Researchers examine sample unfolded proteins, measuring their properties to make predictions about the folding process.
The study of intermediates in the folding pathway provides insights into the molecular details of protein folding within the cell, helping us understand how a protein’s sequence determines its final shape and how it can be facilitated or catalyzed for proper folding.
An Introduction to Protein Folding
Protein folding refers to the physical process by which a linear polypeptide chain, composed of a specific sequence of amino acids, folds into a unique three-dimensional (3D) structure. This 3D structure, known as the native state or tertiary structure, is essential for the protein to perform its biological function.
The study of protein folding is enormously important, as it provides critical insights into:
- Protein function: A protein’s function directly depends on its 3D structure. Hence, understanding folding gives insights into protein function.
- Protein structure prediction: Predicting a protein’s structure from its amino acid sequence remains a key challenge. Advances in understanding folding may improve structure prediction.
- Protein design: Designing proteins with novel structures and functions relies on applying the principles of folding.
- Disease: Misfolded proteins are linked to many diseases like Alzheimer’s and Parkinson’s. Comprehending folding can aid disease understanding and drug design.
Thus, protein folding lies at the heart of fields like structural biology, biotechnology, and medicine. But how exactly do polypeptide chains fold into such precise 3D architectures? This question has puzzled scientists for decades.
The Basic Study of Protein folding and Structure
Protein folding is a process by which a polypeptide chain folds to become a biologically active protein in its native 3D structure. To appreciate the protein folding problem, we must first understand some basics of protein structure:
a. Amino Acids and Polypeptide Chains
Proteins are constructed from linear chains of amino acids linked by peptide bonds. There are 20 common amino acids in proteins, each with a central carbon called the α-carbon, an amino group, a carboxyl group, a hydrogen atom, and a variable R group (or side chain) that differs between amino acids.
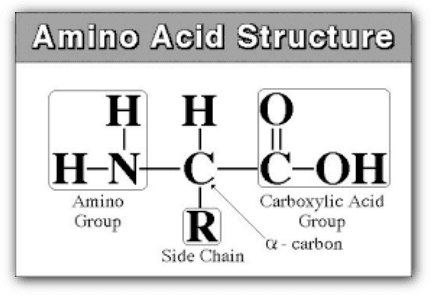
During protein synthesis, amino acids are linked into long chains called polypeptide chains. The sequence of amino acids is determined by the genetic code and is called the primary structure of the protein.
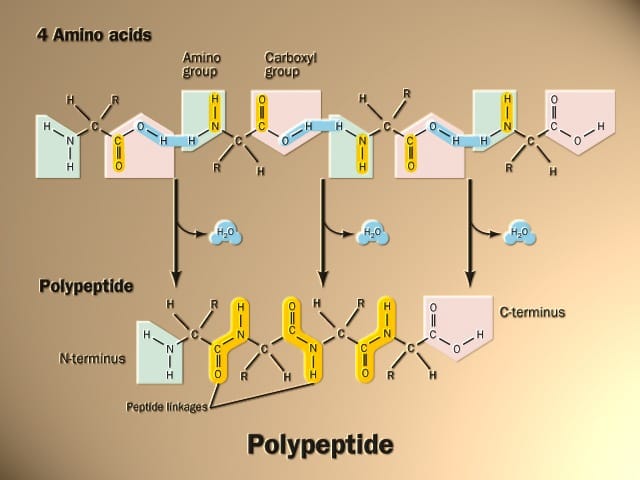
The folding and unfolding of proteins is a dynamic process that is critical to many aspects of protein function and regulation. Correct folding allows a polypeptide chain to adopt its functional three-dimensional structure, while improper folding can lead to the aggregation of misfolded proteins, which is linked to many human diseases.
b. Levels of Protein Structure
Proteins have four levels of structural organization that determine their overall architecture and 3D shape:
Primary Structure
- The primary structure of a protein refers to its amino acid sequence. It is defined by the gene sequence that encodes the protein.
- The specific order of amino acids gives each protein its unique identity.
- Altering the primary structure by even a single amino acid mutation can significantly affects the structure of protein and function.
Secondary Structure
- The most common secondary structure of Proteins are the α-helix and β-sheet conformations formed by hydrogen bonding between backbone atoms.
- α-helices are spiral structures where the protein backbone twists into a right-handed coil stabilized by hydrogen bonds.
- β-sheets consist of extended protein strands connected laterally by backbone hydrogen bonds. β-sheets can be parallel or anti-parallel.
- Secondary structures form sub-units that pack together to give tertiary structure.
Tertiary Structure
- The tertiary structure of proteins refers to the 3D shape of the protein formed by folding of the polypeptide chain.
- It is stabilized by interactions between amino acid R groups (side chains).
- The pattern and architecture of α-helices and β-sheets give each protein a unique tertiary structure.
- Small proteins may contain just one domain while larger proteins have multiple domains in their tertiary structure.
Quaternary Structure
- Quaternary structure arises when multiple protein subunits assemble to form a larger complex.
- The arrangement and interactions between subunits contribute to the quaternary structure.
- Hemoglobin with its 4 subunits is a classic example of a protein with quaternary structure.
In summary, the different levels of protein structure organize from primary sequence to complex 3D shape, guiding protein folding, function and interactions.
c. The Significance of Protein Shape
A protein’s specific 3D shape allows it to interact with other molecules in the cell and perform functions like:
- Enzymatic catalysis: Enzymes have precise active sites that catalyze biochemical reactions.
- Molecular recognition: Proteins like antibodies bind specific targets like antigens.
- Molecular transport: Transport proteins ferry molecules across cell membranes.
- Cell signaling: Signaling proteins relay signals between cells.
- Mechanical work: Structural and motor proteins perform mechanical work like muscle contraction.
Even small structural changes can severely affect a protein’s function. This highlights the importance of the protein folding problem.
Co-translational protein folding
Co-translational protein folding is a fascinating and intricate biological process that plays a crucial role in the proper functioning of cells. This phenomenon can be broken down into several key points:
- Simultaneous Translation and Folding: Co-translational protein folding occurs while a protein is still being synthesized by ribosomes. As the protein’s amino acid chain is being assembled, it begins to adopt a three-dimensional structure.
- Preventing Misfolding: The co-translational process helps to prevent the misfolding of proteins. Misfolded proteins can lead to diseases such as Alzheimer’s and Parkinson’s, making this a critical quality control mechanism.
- Chaperone Proteins: Chaperone proteins are instrumental in guiding the folding process. They interact with the nascent protein chain to assist it in attaining its correct conformation, shielding it from premature interactions that may lead to misfolding.
- Efficiency and Speed: Co-translational folding is highly efficient and rapid. It minimizes the chances of intermediates that might be prone to aggregation and enhances the protein’s functional reliability.
- Post-Translational Modifications: While co-translational folding covers the basics, some proteins may require additional post-translational modifications to achieve their final functional form. These modifications can occur after translation has been completed.
In summary, co-translational protein folding is a dynamic process that ensures the correct structure and function of proteins. It showcases the remarkable coordination between ribosomes, chaperone proteins, and cellular machinery, all of which are essential for maintaining the integrity of cellular processes.
The Protein Folding pathway and its Problem
Protein folding refers to the physical process by which a polypeptide chain acquires precise 3D structure. This occurs through conformational changes driven by thermodynamic forces.
Understanding how linear polypeptide chains fold into 3D shapes is a complex, multidisciplinary problem with several facets, including:
- What forces drive folding?
- What pathways do proteins follow to reach folded state?
- Can we predict a protein’s structure from its sequence?
- How do proteins fold so quickly?
- How do proteins misfold, and what are the consequences?
These questions have challenged researchers for over 50 years. Let’s examine some key aspects of this problem:
a. Levinthal’s Paradox
In 1968, Cyrus Levinthal noted an apparent paradox: if a 100 amino acid protein sampled all possible conformations sequentially before reaching its folded state, it would take longer than the age of the universe to fold! This is because the number of possible conformations is astronomical.
But most small proteins fold within milliseconds. How? The answer is that proteins do not sample all conformations, but follow directed folding pathways guided by thermodynamic forces. Resolving Levinthal’s paradox was an early triumph in folding research.
b. The Challenge of Protein Structure Prediction
A key goal in structural biology is to predict a protein’s 3D structure from its amino acid sequence. But this remains exceptionally challenging, especially for large multi-domain proteins.
While progress has been made using comparative modeling and AI, protein structure prediction remains unsolved. Advances in folding research may pave the way for tackling this grand challenge.
Nature and Regulation of Protein Folding on the Ribosome
Protein folding on the ribosome is a complex and highly regulated process that plays a crucial role in the synthesis of functional proteins. The process of folding begins co-translationally, as the structure of a protein starts to take shape while the chain is still being synthesized on the ribosome.
The ribosome is the cellular machinery responsible for translating the genetic code in messenger RNA (mRNA) into a polypeptide chain, which subsequently needs to fold into its native three-dimensional structure to become a functional protein. Here’s an overview of the nature and regulation of protein folding on the ribosome:
1. Nascent Polypeptide Chain Formation: As the ribosome moves along the mRNA, it synthesizes a nascent polypeptide chain by linking amino acids together according to the genetic code. This nascent chain is initially in an extended, unstructured conformation.
2. Co-translational Folding: Protein folding on the ribosome is primarily co-translational, meaning that the folding process occurs as the polypeptide chain is being synthesized. The growing polypeptide chain emerges from the ribosome’s exit tunnel, and various folding events can start even before the entire chain is synthesized.
3. Chaperone Proteins: Chaperone proteins, such as trigger factor in bacteria and signal recognition particle (SRP) in eukaryotes, assist in co-translational protein folding. These chaperones help prevent misfolding, aggregation, or premature interactions with other cellular components.
4. Ribosome Exit Tunnel: The ribosome exit tunnel is a channel through which the nascent polypeptide chain emerges from the ribosome. This tunnel is an important environment for initial folding and can influence the folding pathway of the emerging protein.
5. Domain Formation: Some proteins consist of multiple domains, and domain formation can occur as individual segments of the polypeptide chain are synthesized. This process can be driven by hydrophobic interactions, disulfide bond formation, or other specific interactions between amino acid residues.
6. Regulatory Factors: Ribosome-associated factors and specific RNA sequences can regulate the speed of translation and the co-translational folding process. Slower translation rates can facilitate more extensive folding, while faster translation can lead to less complete folding.
7. Post-translational Folding: While most of the folding occurs during translation, post-translational folding events also play a role in achieving the correct protein conformation. This can involve further modification, cleavage, or the assistance of additional chaperones in the cytoplasm.
8. Quality Control Mechanisms: Cells have quality control mechanisms to ensure that only properly folded proteins are released. If a protein does not fold correctly, it can be targeted for degradation by the proteasome or other protein degradation pathways.
9. Regulatory Feedback: In some cases, the ribosome’s translation rate and co-translational folding process are regulated in response to cellular conditions, including the availability of specific cofactors, chaperones, or environmental stressors.
The kinetics of protein folding determine how rapidly the folding process takes place once the polypeptide chain is synthesized.
Small single-domain proteins may fold in microseconds, while larger multi-domain proteins can take seconds to minutes to achieve their fully folded native state.
Co-translational protein folding begins while the protein is still being synthesized by the ribosome. The initial co-translational folding events help guide the downstream folding process.
In summary, protein folding on the ribosome is a dynamic and regulated process that involves co-translational folding, chaperone assistance, domain formation, and quality control mechanisms.
It is critical for ensuring that the nascent polypeptide chain adopts its functional three-dimensional structure, which is essential for the protein’s proper biological function.
Protein Folding in the Endoplasmic Reticulum
- Protein folding in the endoplasmic reticulum (ER) is crucial for the proper functioning of secretory and membrane proteins in eukaryotic cells.
- The endoplasmic reticulum, a membranous organelle, is central to protein synthesis and folding.
- Ribosomes on the rough ER synthesize proteins that enter the ER lumen.
- Folding and post-translational modifications occur in the ER, with chaperone proteins like BiP guiding the process.
- Disulfide bond formation, glycosylation, and other modifications enhance protein stability and functionality.
- Quality control mechanisms ensure only properly folded proteins exit to their target destinations, like the Golgi apparatus.
- Proper protein folding is vital for cellular homeostasis, preventing cellular stress responses like the unfolded protein response (UPR).
- The UPR, if prolonged, can lead to cell dysfunction and disease.
- In summary, the endoplasmic reticulum plays a central role in precise protein folding.
- Protein folding ensures identical amino acid sequences fold into the exact same 3D structures, enabling specific functions.
- In the endoplasmic reticulum, protein disulfide isomerase is a key enzyme involved in protein folding by catalyzing disulfide bond formation and rearrangement steps.
- The redox conditions in the ER also promote correct disulfide bond formation during folding in the endoplasmic reticulum.
Forces Driving Protein Folding
Proper folding is essential for proteins to achieve their functional conformations. A variety of intracellular factors assist in the folding process and prevent misfolding or aggregation. Understanding the forces that drive correct protein folding and the techniques for studying protein folding dynamics remain key areas of research, as this knowledge can shed light on disease mechanisms linked to misfolding.
What forces guide protein chains to adopt specific compact 3D structures? Four key interactions stabilize protein tertiary structures:
a. Hydrophobic Interactions
The dominant forces driving protein folding are hydrophobic interactions between nonpolar amino acids. To reduce unfavorable exposure to water, hydrophobic groups bury in the protein core, driving folding.
b. Hydrogen Bonding
Backbone hydrogen bonding between N-H and C=O groups stabilizes secondary structures like α-helices and β-sheets. This pre-structures the chain for subsequent folding.
c. Van der Waals Forces
These transient electrical forces between nearby atoms help pack protein interiors densely. This removes empty space and strengthens tertiary structures.
d. Electrostatic Interactions
Ionic interactions between charged amino acids help orient protein domains and stabilize tertiary structures. Disulfide bridges between cysteine residues also constrain structures.
Furthermore, molecular chaperones can help guide folding by preventing misfolding and aggregation. Hence, a variety of noncovalent forces stabilize protein folds. Understanding these has shed light on the folding process.
Protein Misfolding and Disease
Folding and misfolding are in constant competition within the complex cellular environment. The protein may be kinetically trapped in misfolded states that cannot easily reach the functional native structure without assistance.
Studying the folding of some proteins facilitates the proper folding of proteins within the native state of the protein by measuring the role in protein folding, which is determined by the description of protein folding, while other proteins are involved in the folding of some proteins, and a sample of unfolded protein can help unfold the protein before it reaches its native state, ensuring that each protein is able to fold correctly.
While most proteins fold reliably into functional structures, molecular mishaps can cause misfolding. This can have severe cellular consequences:
a. Protein Misfolding and Amyloid Formation
When proteins misfold, they often aggregate into long insoluble fibrils called amyloids. Amyloid formation is linked to dozens of diseases, including:
- Alzheimer’s disease: Amyloid plaques formed by misfolded amyloid beta peptide.
- Parkinson’s disease: Intracellular Lewy bodies contain misfolded α-synuclein protein.
- Prion diseases: Misfolded prion protein aggregates into neurotoxic oligomers and fibrils.
b. Loss and Gain of Function
Misfolded proteins can either lose normal function or gain toxic functions. For example:
- Cystic fibrosis occurs due to loss of function of misfolded ΔF508 mutant CFTR protein.
- Prion replication occurs due to gain of function of misfolded prion protein.
c. Therapeutic Implications
Elucidating how and why proteins misfold is vital for developing treatments for misfolding diseases like Alzheimer’s, cystic fibrosis and prion disorders. Drugs that reverse protein misfolding could effectively treat such conditions.
The correct configuration of the protein in its native state is essential for its proper function within the cell. Diseases caused by protein misfolding highlight the importance of factors that catalyze protein folding and prevent misfolded proteins from aggregating.
Experimental Techniques for Studying Protein Folding
The sequence of a protein determines its final shape and structure. Each part and portion of the protein folds into a specific configuration to achieve the native structure of the protein. The kinetics of protein folding determine how rapidly the process takes place. Protein folding and misfolding are in constant competition as the protein tries to achieve its functional form.
Researchers employ a variety of biophysical techniques to study protein folding:
a. X-Ray Crystallography
This technique reveals protein structures at atomic resolution. Researchers can visualize structural changes between unfolded and folded states. Over 177,000 protein structures in the Protein Data Bank were solved using X-ray crystallography.
b. NMR Spectroscopy
Nuclear magnetic resonance (NMR) spectroscopy monitors protein structure and dynamics in solution. NMR can probe transient partially folded states during folding.
c. Circular Dichroism Spectroscopy
This technique measures differences in the absorption of left and right circularly polarized light by proteins. It provides information on protein secondary structure and folding.
c. Fluorescence Spectroscopy
Intrinsic tryptophan fluorescence or fluorescent tags are used to monitor conformational changes during folding.
d. Mass Spectrometry
Mass spectrometry (MS) determines the mass-to-charge ratio of protein ions. Used alongside protein labeling, MS reveals protein folding kinetics and pathways.
The kinetics of protein folding determine how rapidly the process takes place. Small proteins may fold in microseconds, while larger proteins can take seconds to minutes to achieve their final folded forms.
Either only a part of the protein or the whole complete polypeptide chain is found in this disorder. Therefore, investigating only some parts of the proteins would not help summarize the flexibility of the protein.
The term “conditionally disordered” means the disorder of proteins may happen under some certain conditions and may not happen under other conditions.
e. Cryo-Electron Microscopy
Cryo-EM images proteins in near-native solution states. Combined with computational image processing, it achieves near atomic resolution, helping visualize folding intermediates.
Computational Approaches to Study Protein Folding
Computational prediction of protein tertiary structure continues to advance our understanding of complex biomolecules. Alongside experiments, computational techniques help study folding:
a. Molecular Dynamics Simulations
Molecular dynamics (MD) simulations compute the time-dependent behavior of proteins in atomic detail. This reveals transient folding intermediates hard to capture experimentally.
b. Monte Carlo Simulations
Monte Carlo (MC) methods use repeated random sampling to model folding processes. This helps estimate thermodynamic properties driving folding.
c. Rosetta Folding Software
The Rosetta software suite incorporates modeling, simulation and AI for de novo protein structure prediction and design.
d. Homology Modeling
Known protein structures provide templates to computationally model related protein structures. Useful when no experimental structure is available.
f. Artificial Intelligence for Structure Prediction
Deep learning holds promise for learning patterns from big data to improve protein structure and function prediction. For example, DeepMind’s AlphaFold2 leverages deep learning and achieves over 90% structure prediction accuracy.
Notable Discoveries and Milestones in Folding of a protein
Key discoveries have gradually unravelled the mysteries of polypeptide folding:
a. Anfinsen’s Experiment on Ribonuclease Folding
In 1961, Christian Anfinsen showed that ribonuclease could refold into its native state after denaturation, demonstrating that all information required for folding is encoded in the protein’s primary sequence. This pioneering finding earned Anfinsen the 1972 Nobel Prize.
When a protein is denatured, it unfolds and loses its functional three-dimensional structure. Refolding involves the protein finding its way back to the fully folded native state.
While the main principles of folding are universal, each protein sequence has its own optimal folding trajectory that takes place along the energetically most favorable pathway.
b. Elucidation of Disulfide Bond Formation
The oxidative folding of proteins like ribonuclease in the endoplasmic reticulum was elucidated in the 1960s. This revealed how disulfide bonds help guide folding. Formation of protein disulfide bonds often occurs concurrently with folding and helps stabilize the native protein structure.
c. Discovery of Chaperonins
Chaperonins are protein complexes that assist folding in cells. Their discovery in the late 1980s shed light on how molecular chaperones prevent misfolding. Molecular chaperones within the cell help guide the process of folding, prevent aggregation of misfolded proteins, and promote proper refolding of denatured proteins.
While some proteins can spontaneously fold, others require the involvement of molecular chaperones to prevent incorrect folding and aggregation. These chaperones play a key role in stabilizing intermediates during the folding and assembly process.
d. Advances in Simulations and Computer Algorithms
From simple lattice models in the 1970s to today’s sophisticated molecular dynamics and AI algorithms, computational methods have revolutionized how folding is studied.
e. Crowdsourced Protein Folding: Foldit
The Foldit computer game lets players predict protein structures by folding virtual chains. Showcases the power of crowdsourcing difficult scientific problems.
f. Growth of the Protein Data Bank (PDB)
The PDB archive of experimentally determined protein structures has been pivotal for structure prediction and computational modeling of folding. The PDB currently contains over 177,000 protein structures.
Recent advances in AI makes gigantic leap in solving protein structures. In 2020, the AI system AlphaFold2 developed by DeepMind made a tremendous breakthrough by accurately predicting the structures of many types of proteins – a feat considered impossible just a few years ago. This leap in solving protein structures computationally could revolutionize our understanding of the protein folding process.
The Future of Protein Folding Research
The development of human body is needed to be parallel with the development of protein. But protein contains so many mysteries that we did not discovery yet. For example, that is protein folding.
Recent technological advances are tremendously exciting for protein folding research:
- Cryo-electron microscopy (cryo-EM) can now visualize protein folding intermediates at near-atomic resolution.
- Room temperature X-ray crystallography allows dynamic structural snapshots of proteins in action.
- Single-molecule techniques probe protein motions in real-time with high spatial and temporal precision.
- Massively parallel supercomputing enables large-scale atomistic simulations of complex folding phenomena.
- AI and deep learning hold promise for accurately predicting protein structure from sequence.
These developments will likely yield profound insights into protein folding dynamics and lead to a new era of protein structure prediction and design.
Key open questions remain to be addressed:
- How do proteins fold so quickly? The Levinthal paradox is still not fully resolved.
- Can we predict protein structure ab initio from sequence alone? This remains extremely challenging.
- What non-native states are populated during folding? Their characterization remains limited.
- How exactly does mutation alter folding pathways and cause misfolding diseases? Molecular details are sparse.
- Can drugs be designed to prevent protein misfolding? Progress remains slow despite huge potential.
As we seek to overcome these challenges, the payoffs for medicine, biotechnology and basic science will be transformative.
Frequently Asked Questions (FAQs)
What is protein folding?
Protein folding is the physical process by which a protein chain acquires a precise three-dimensional structure essential for its biological function. Folding is driven by a variety of chemical interactions and transforms disordered polypeptide chains into compact, functional protein molecules.
Why is protein folding important?
Correct protein folding is crucial for building functional proteins. Misfolded proteins often aggregate into toxic deposits linked to diseases like Alzheimer’s and Parkinson’s. Understanding protein folding provides insights into protein structure, function and design.
What forces drive protein folding?
The main forces driving protein folding are hydrophobic interactions, hydrogen bonding, van der Waals forces and electrostatic interactions. Hydrophobic groups bury in the protein core to avoid water contact, while hydrogen bonds stabilize secondary structures like α-helices.
What is Levinthal’s paradox in protein folding?
Levinthal noted that proteins seem to sample all possible conformations to find the folded state, which would take astronomically long times. But proteins fold within milliseconds. This is because folding is guided by thermodynamics along directed pathways, not random sampling.
What are some diseases associated with protein misfolding?
Many diseases involve protein misfolding and aggregation, including Alzheimer’s (amyloid beta aggregates), Parkinson’s (α-synuclein aggregates), cystic fibrosis (mutant CFTR), prion disorders (prion protein) and amyotrophic lateral sclerosis (SOD1 aggregates).
Final Words
In summary, protein folding is a complex molecular phenomenon vital to all aspects of protein structure, function and dysfunction.
Understanding how amino acid chains reliably fold into precise 3D architectures despite astronomical possibilities has puzzled researchers for over 50 years.
Key insights into the forces driving folding have been obtained through an interplay of elegant biophysical experiments and computational modeling.
Despite significant progress, protein folding continues to be an immensely exciting field, with open questions remaining to be addressed.
Advances promise to revolutionize our ability to comprehend, model and manipulate the molecular basis of life.
Keynote points:
- Researchers study protein folding by comparing properties of sample proteins under folded and unfolded conditions to predict folding mechanisms.
- Some proteins, like RNase A, can spontaneously fold in vitro, while others require chaperones to prevent incorrect folding.
- Molecular chaperones interact with intermediates, stabilizing them and aiding proper protein folding in cells.
- Protein misfolding-related diseases emphasize the importance of factors catalyzing folding and preventing misfolded protein aggregation.
- The theory of protein folding explains how a protein’s sequence dictates its final three-dimensional shape.
- Researchers explore protein folding stages and chaperone roles in mediating the process, particularly focusing on transition states and intermediates.
- Protein folding within cells follows directed pathways driven by thermodynamic principles but is influenced by complex interactions due to the crowded cellular environment.
- Further research is needed to understand protein folding dynamics within living cells, and it can inform disease-related studies.
- Discovering the roles of cellular factors in protein folding can enhance our knowledge of misfolding-related diseases and potentially improve protein folding within cells through increased chaperone activity.
- Insights into molecular details of folding within the complex intracellular environment can aid these efforts.
- The folding rate determines how rapidly the process of protein folding takes place following synthesis of the polypeptide chain.
- Folding can begin co-translationally while the protein is still being synthesized on the ribosome.
- Molecular chaperones within the cell regulate various aspects of protein folding by preventing aggregation and catalyzing correct folding of the polypeptide into its native shape.
- Recent advances in AI have enabled dramatic leaps in computationally predicting protein structures from sequence, shedding light on the determinants of folding.
- Researchers study protein folding by measuring properties of sample proteins under unfolded and folded conditions to elucidate the protein folding reactions involved.
- Comparison of folded and unfolded states allows prediction of the transitions that take place as the protein reaches its native configuration.
- Within cells, biological factors like chaperones facilitate correct protein folding while also being able to unfold misfolded proteins.
Discover more from Biochemistry Den
Subscribe to get the latest posts sent to your email.





