Phenylketonuria (PKU): Definition, Causes, Symptoms and Treatment
In the United States, about 1 in 10,000 to 15,000 babies is born with phenylketonuria (PKU) every year. This inherited metabolic disorder occurs in all ethnic groups but is more common in people of Northern European ancestry and some Native American populations.
What is phenylketonuria?
Phenylketonuria (PKU) is an inherited metabolic disorder in which the body cannot properly metabolize the amino acid phenylalanine.
In simple terms, phenylketonuria is a genetic disease where a defective enzyme prevents the normal breakdown of phenylalanine, leading to its accumulation in the blood and tissues.
Phenylalanine is an essential amino acid present in high‑protein foods and in some artificial sweeteners such as aspartame, which is why products containing aspartame often carry the warning “contains phenylalanine” for people with PKU. Amino acids, including phenylalanine, are the basic building blocks of proteins in the body.
A short phenylketonuria definition you can remember for exams is
Phenylketonuria is an autosomal recessive inborn error of metabolism caused by a deficiency of the enzyme phenylalanine hydroxylase, leading to toxic accumulation of phenylalanine and its metabolites.
Because PKU is a recessive genetic disorder, a person must inherit two defective copies of the gene (one from each parent) to develop the disease.
People who carry only one defective gene copy are called carriers of phenylketonuria; they usually have no symptoms but can pass the gene to their children.
What causes PKU?
The primary cause of PKU is a mutation in the PAH gene, which codes for the enzyme phenylalanine hydroxylase.
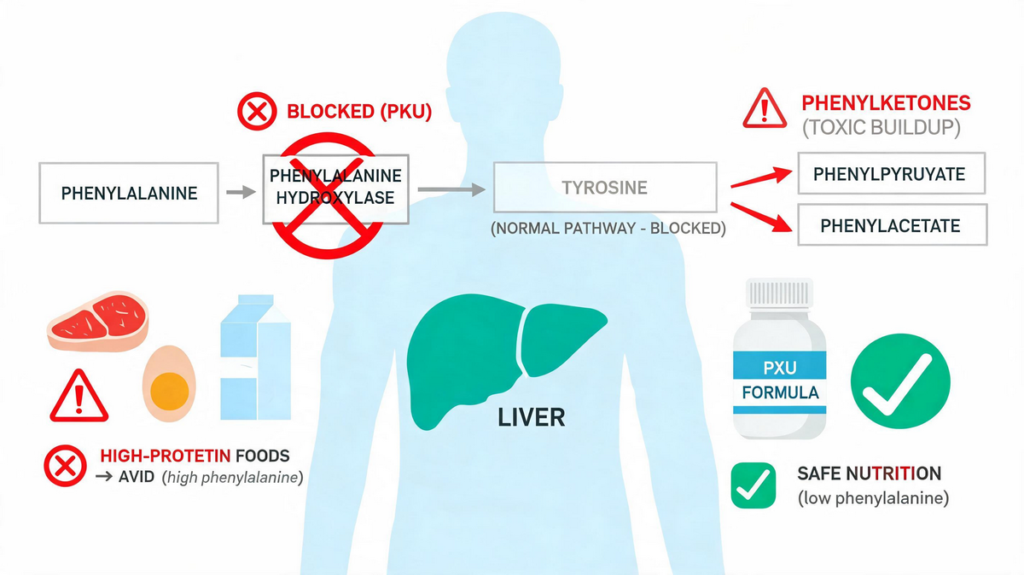
This enzyme is required to convert phenylalanine to tyrosine in the liver; when it is absent or deficient, phenylalanine and its abnormal metabolites accumulate.
So, phenylketonuria is caused by:
- An autosomal recessive mutation in the PAH gene
- Markedly reduced or absent activity of the phenylalanine hydroxylase enzyme
- Subsequent buildup of phenylalanine in blood, brain, and other tissues
From a biochemistry of phenylketonuria point of view:
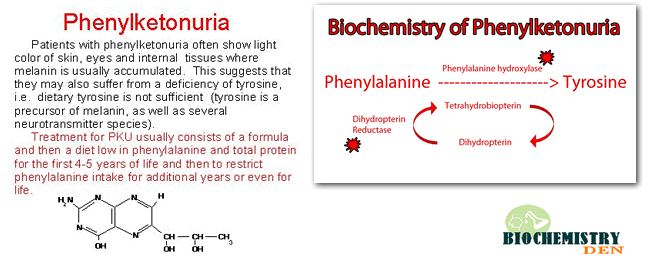
- Normally, phenylalanine is converted to tyrosine via phenylalanine hydroxylase, using tetrahydrobiopterin (BH₄) as a cofactor.
- In PKU, this phenylketonuria reaction is blocked.
- Excess phenylalanine is converted into phenylpyruvate, phenylacetate, and phenyllactate, which appear in urine and give the condition its name (“phenyl‑ketone‑uria”).
Aspartame and PKU: Aspartame is a low‑calorie artificial sweetener that contains phenylalanine, so individuals with PKU or their parents often ask why aspartame labels warn people with phenylketonuria. The reason is that consuming aspartame significantly increases phenylalanine intake and can be dangerous for people with PKU.
How to tell if your baby has PKU?
How is phenylketonuria diagnosed? Almost all countries now perform newborn screening for PKU. This screening detects PKU very early, often before any phenylketonuria symptoms appear.
Shortly after birth (usually within the first 24–72 hours), a health professional collects a few drops of blood from the baby’s heel onto special filter paper. The laboratory measures phenylalanine levels and reports whether the newborn screen is normal or suggests PKU disease.
If the newborn screening test is abnormal:
- The baby will need confirmatory diagnostic tests to differentiate true PKU from false‑positive results.
- Additional blood tests measure phenylalanine and tyrosine levels and may include genetic testing for PAH mutations.
Because very early samples (less than 24 hours after birth) can miss PKU, many guidelines recommend repeating the test at about 1–2 weeks of age if the initial sample was collected too early.
This is why parents sometimes see recommendations like “If the first PKU test was done within 24 hours, repeat at 7–14 days.”
Phenylketonuria symptoms
Babies with PKU usually look normal at birth and for the first few months of life. Without diagnosis and treatment, however, the progressive accumulation of phenylalanine leads to typical phenylketonuria symptoms beginning around 6 months of age.
Important clinical features of PKU include:
- Jerky movements or tremors in the arms and legs
- Very fair skin, hair, and eye colour due to reduced melanin synthesis
- A characteristic musty or “mouse‑like” body odour
- Recurrent skin rashes (often eczema‑like)
- Small head size (microcephaly) as the brain fails to develop normally
- Developmental delay: sitting, crawling, or walking later than expected
- Loss of interest in the surroundings and poor social interaction
- Delayed speech and impaired social skills
- Progressive intellectual disability if untreated
- Behavioural problems such as hyperactivity, irritability, or autistic‑like features
- Seizures in more severe, untreated cases
These manifestations explain why exam questions often ask the following:
- What are the symptoms of phenylketonuria?
- Phenylketonuria characteristics / features
- What happens in phenylketonuria if it is not treated?
Maternal phenylketonuria: Women with poorly controlled PKU during pregnancy may have babies with congenital heart defects, microcephaly, and growth retardation, even if the baby does not have PKU. This is called maternal PKU syndrome and highlights the importance of strict dietary control before and during pregnancy.
Phenylketonuria treatment
If your baby has PKU, what kind of treatment do you need?
There is no complete cure for phenylketonuria, but early diagnosis and lifelong management can prevent almost all neurological damage.
The cornerstone of PKU treatment is dietary restriction of phenylalanine with regular monitoring of blood phenylalanine levels.
Key principles of treatment of phenylketonuria:
- Early and strict dietary management
- Start a low‑phenylalanine diet ideally within the first 7–10 days of life once PKU is confirmed.
- Use a special PKU‑specific formula that contains all essential amino acids except phenylalanine, along with vitamins and minerals.
- Carefully control the amount of natural protein (phenylalanine) from breast milk or standard formula in consultation with a metabolic specialist.
- Allowable and restricted foods Babies and children with PKU can eat the following:
- Fruits and most vegetables Some low‑protein grains such as special low‑protein bread, pasta, and cereals Measured amounts of specific low‑protein products designed for PKU
- Milk, cheese, ice cream, and other dairy products
- Eggs
- Meat and poultry
- Fish
- Nuts and seeds
- Legumes such as beans and lentils
- Common foods or beverages containing aspartame (diet soft drinks, sugar‑free gum, some protein bars), because aspartame is a source of phenylalanine
- Lifelong management
- Modern guidelines recommend that individuals with PKU continue their low‑phenylalanine diet for life, not just during childhood.
- Blood phenylalanine levels should be monitored regularly, more frequently in infancy and early childhood, and at longer intervals later in life.
- Women with PKU must achieve tight metabolic control before conception and throughout pregnancy to prevent maternal PKU syndrome in the fetus.
- Drug therapy
- Sapropterin dihydrochloride is a synthetic form of tetrahydrobiopterin (BH₄) and can help some patients with mild PKU who still have residual enzyme activity.
- When responsive, sapropterin lowers blood phenylalanine levels and may allow a more liberal diet, but it does not replace the need for careful dietary management.
- Newer therapies, such as pegvaliase (an enzyme substitution therapy) for adults with uncontrolled PKU, are emerging options in some countries and are usually reserved for specialized centers.
The accurate answer is that PKU is best treated by lifelong restriction of dietary phenylalanine plus, in selected patients, pharmacological therapies, which together prevent brain damage and allow normal growth and development.
FAQ – People Also Ask Optimized
What is phenylketonuria in simple words?
Phenylketonuria, or PKU, is a rare genetic disorder in which the body cannot break down an amino acid called phenylalanine. When phenylalanine builds up, it can damage the developing brain and nervous system. With early diagnosis through newborn screening and a special low‑phenylalanine diet, most children with PKU can grow, learn, and live a normal, healthy life.
Phenylketonuria is an example of which disorder?
Phenylketonuria is an example of an autosomal recessive inborn error of metabolism involving the amino acid phenylalanine. In this disorder, a defective gene reduces the activity of the enzyme phenylalanine hydroxylase, which normally converts phenylalanine to tyrosine. Because the enzyme does not work properly, phenylalanine and its toxic metabolites accumulate, especially in the brain, causing developmental delay and intellectual disability if PKU is not treated early.
What is PKU disease caused by?
PKU disease is caused by mutations in the PAH gene, which provides instructions for making the enzyme phenylalanine hydroxylase. This enzyme normally helps convert phenylalanine into another amino acid, tyrosine, in the liver. When the enzyme is missing or severely reduced, phenylalanine builds up to harmful levels in the blood and brain. The condition is inherited in an autosomal recessive pattern, so a child must receive the faulty gene from both parents to develop PKU.
Can PKU be cured?
PKU cannot be completely cured at present, because the underlying genetic change cannot yet be permanently reversed in routine clinical practice. However, PKU can be very effectively managed with early diagnosis and lifelong treatment. A strict low‑phenylalanine diet, PKU‑specific medical formulas, regular blood monitoring, and, in some patients, medications like sapropterin or enzyme therapy can keep phenylalanine at safe levels. With good control, many people with PKU lead active, independent lives.
What is the relationship between aspartame and PKU?
Aspartame is an artificial sweetener used in many sugar‑free drinks, chewing gums, and diet foods. When the body breaks down aspartame, it releases phenylalanine, the same amino acid that people with PKU cannot process properly. Even small extra amounts of phenylalanine from aspartame can raise blood levels and pose a risk to the brain in individuals with PKU. That is why products containing aspartame carry a special warning, and people with phenylketonuria are advised to strictly avoid aspartame‑containing foods and beverages.
What are the early signs of phenylketonuria in babies?
Early signs of phenylketonuria in babies are often subtle and may not appear immediately after birth. Over the first few months, parents might notice poor feeding, vomiting, unusual irritability, or a musty odor from the baby’s skin or urine. As phenylalanine continues to build up, delayed milestones, small head size, lighter skin and hair, and seizures may develop. Newborn screening is crucial because it detects PKU before these early symptoms become obvious.
How is PKU inherited from parents?
PKU is inherited in an autosomal recessive pattern, which means a child must receive one faulty PAH gene from each parent to develop the disorder. Parents who each carry a single mutated gene are usually healthy but have a 25% chance in every pregnancy of having a child with PKU. They also have a 50% chance of having a carrier child and a 25% chance of a child with no mutation. Genetic counseling helps families understand these inheritance risks and plan future pregnancies.
What happens if phenylketonuria is not treated?
If phenylketonuria is not diagnosed and treated early, phenylalanine levels in the blood and brain rise to toxic levels. Over time, this causes irreversible damage to the developing nervous system. Untreated children typically develop severe intellectual disabilities, behavioral problems, seizures, and microcephaly, and many have a characteristic musty body odor and very fair coloring. Because brain damage is permanent, timely newborn screening and immediate dietary treatment are essential to prevent these serious outcomes.
What foods should people with PKU avoid?
People with PKU should avoid foods that are naturally high in protein and phenylalanine, because these can quickly raise blood phenylalanine levels. This includes meat, fish, eggs, regular dairy products, soy products, nuts, and most legumes. Many baked goods, chocolates, and processed snacks are also too high in protein. In addition, they must completely avoid foods and drinks containing aspartame, a sweetener that releases phenylalanine. A dietitian helps design a safe meal plan with special low‑protein products.
Can adults have symptoms of PKU if the diet is relaxed?
Yes, adults with PKU can develop symptoms again if they relax their low‑phenylalanine diet too much. High phenylalanine levels in adulthood are linked to problems such as difficulty concentrating, memory issues, mood changes, anxiety, depression, and headaches. Some people also report decreased executive function and slower information processing. Because of these potential effects, current recommendations encourage lifelong dietary control and regular monitoring, rather than stopping the PKU diet after childhood.
Summary / Final words
Phenylketonuria (PKU) is a classic example of how early diagnosis and simple dietary changes can completely transform a child’s future.
When newborn screening detects PKU and treatment starts quickly, most children can avoid brain damage and lead healthy, productive lives.
Lifelong attention to diet, regular monitoring, and awareness of hidden sources of phenylalanine, such as aspartame, remain essential.
With proper support from healthcare teams and family, PKU is a manageable condition rather than a life‑limiting disease.
Discover more from Biochemistry Den
Subscribe to get the latest posts sent to your email.